Sequencing throughput is only going to increase, and it is a worthwhile exercise to inspect one's own bioinformatics set up to ensure readiness and modularity for future technologies and data. Use an Adapter design pattern now to avoid major headaches later!
One of the most agonising aspects of bioinformatics pipeline development and maintenance is having to deal with the replacement of obsoleted or superseded software packages. As pipelines grow and fork like Lernaean Hydrae (Fig. 1), the number of cheeky file-format conversion scripts quickly spirals out of control at the same inverse rate as the enthusiasm with which the system is documented.
At research institutions and, as I gleaned recently at a conference, pre-competitive facilities in biotech and pharma, this usually means that the bioinformaticians in charge of these pipelines are suddenly the only people aware of how everything works. The majority of their day is spent ensuring the pipelines behave appropriately, rather than analysing data sets quickly, or even finally getting down to documenting the whole thing. So how do we combat this madness in a way that minimally impacts current processes without stretching the pipeline developers even further?
I am an idealist and a fan of the butterfly effect. I believe that effecting one small positive change upstream triggers massive improvements downstream, and the further upstream the change is, the bigger the improvement is downstream. I also believe in doing as little manual, repetitive work a possible when my job is to make a computer go faster and make better decisions every day. At the Oxford Genomics Centre, we recently switched to a new Laboratory Information Management System (LIMS) and I was tasked with making the transition as smooth as possible. “Transition” refers to the deployment and configuration of the new LIMS, connection to the bioinformatics pipelines, documentation of the whole system, disconnecting the old LIMS, and training and support for both the lab genomics team and the bioinformaticians. Whereas the new LIMS had its own configuration and data exchange format quirks that needed to be overcome in order to make it operational as a standalone piece, the interconnection to other components was made significantly easier by the presence of a Python /Django /PostgreSQL application affectionately called the “tracking database”.
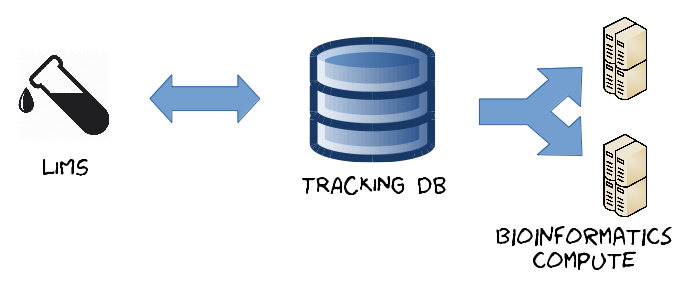
Our downstream bioinformatics pipelines are connected to this application, which copies necessary metadata (flow cells, libraries, Illumina run folder locations etc.) from the LIMS to drive the pipeline scripts. The old LIMS system at that time was temperamental, to say the least, and required frequent reboots and maintenance down-time. A bioinformatics infrastructure needs to have 24-hour reliability, of course, so this Django-based tracking database was built to collect LIMS metadata whilst effectively ring-fencing the computation away from the lab processes (Fig. 2).
I believe that effecting one small positive change upstream triggers massive improvements downstream, and the further upstream the change is, the bigger the improvement is downstream.
This set up meant that the implementation of the new LIMS did not affect the downstream analysis work, and the only major connection I had to build was between the LIMS and the tracking database (data structure interchange, essentially). Serendipitously, we had ended up with a modular, extensible software system that afforded us the luxury of gradually changing a core component such as the LIMS (whilst running the old LIMS in parallel for some time) without significantly impacting the bioinformatics analysis. From the customer's point of view, this meant we could continue to deliver our QC reports and analysis data efficiently, and negative effects and delays due to changing to the new LIMS were greatly minimised.
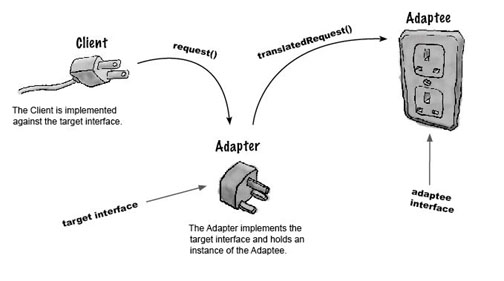
In software design parlance the connection of the LIMS to the tracking database most closely represents the Adapter design pattern, which joins two pieces of code that were not originally designed to work with each other (Fig. 3). The implementation was not seamless of course; I had to obsolete some fields in some of the PostgreSQL tables whilst building new models, functions and API calls to make the tracking database gel with the LIMS as quickly as possible. To support my work, I constructed a web-accessible documentation system on a second server (so as to not clash with the tracking server or the LIMS) using the brilliant Sphinx documentation tool which auto documented most of the new Python, and gave me a private scratch pad to quickly note down website references, code snippets, and caveats in how the system needed to be set up (the second server also presents a web portal to directly interact with the LIMS system for extending the features of the LIMS itself, but this is a topic for another day). Of course, the whole system is now revision controlled with Git.
Sphinx ensures that every piece of code I touch is documented and presentable.
The end result is that the new LIMS system is now fully integrated and drives the bioinformatics through the tracking database. As we grow and incorporate new methods of sequencing, the tracking database can be extended quickly to drive new pipelines or upgrade existing ones. The modular nature of the system also means that each piece can now be moved onto its own redundant hardware set up as we begin to obsolete old computers. New features can be tested rigorously before rolling them out onto the production set up, and mistakes can be quickly rolled back. Sphinx ensures that every piece of code I touch (especially bits that were not originally written by me!) is documented and presentable. Critically, the modularity also means that individual software developers or bioinformaticians could, in theory, build modules without any grey areas in ownership or duplication of work (we are not yet in this rapid software production situation, but it should always be remembered as a future possibility!).
An appliance such as our tracking database might seem like overkill for small labs, but the return on investment is greatly amplified when the system needs to quickly grow or adapt to new sequencing methods, a new LIMS, or even a large conceptual change. In regards to the latter, my next goal for our pipelines is to wrap them using something like the Common Workflow Language (CWL) to further modularise the downstream scripts and code. This will invariably involve extensions to the tracking database to store metadata arising from the CWL (especially software versions and locations on disk), but it is an important exercise for documenting, taming, testing, and rapidly deploying new analysis routines, especially on cluster architectures. Sequencing throughput is only going to increase, and it is a worthwhile exercise to inspect one's own bioinformatics set up to ensure readiness and modularity for future technologies and data. Use an Adapter now to avoid major headaches later!
By Varun Ramraj, D. Phil., Automation & Information Management Developer, Oxford Genomics Centre, Wellcome Trust Centre for Human Genetics, University of Oxford
[tw_callout size="waves-shortcode" text="" callout_style="style2" thumb="" btn_text="Republish the article" color="#37a0d9" btn_url="https://scinote.net/blog/republish/" btn_target="_blank"]
{kind=link}
{kind=link}
{kind=link}